Herpesviridae: Viral Cycle, Capsid Transport, and Cancer Treatment
Introduction

By Michael Gallaher
Herpes is a family of DNA viruses called Herpesviridae. The herpes family of viruses consists of three families of viruses; Alphaherpesviridae, Betaherpesviridae, and Gammaherpesviridae. Among these families are many commonly known viruses that are causative agents of many diseases, such as HSV-1 and HSV-2 for cold sores and genital warts, varicella zoster for chicken pox and shingles, and the Eppstein-Barr virus for mononucleosis. Herpes virsues also tend to have latent, recurring infections in the infected organisms, where the virus remains in some part of the infected organism (Gupta, 2007). Herpes family viruses are incredibly common, with at least 90% of people within the United States having been infected with at least one form of Herpesviridae (CDC, 2006).
Of these viral families, the animal herpes viruses that cause disease are of the Alphaherpesviridae family. The most well known examples of herpes are the herpes simplex viruses, HSV-1 and HSV-2. HSV-1 is the causative pathogen of most oral cold sores, while HSV-2 is the causative agent of genital warts. It is thought that more than 50% of adults in the United States have HSV-1, and that nearly 20% have HSV-2 (CDC, 2010). The widespread infection of the populace makes herpes a good target for medical research. Another promising field of research that has developed more recently is the possibility of using a re-engineered HSV as a treatment vector for other diseases, such as various forms of cancer (Varghese, 2002).
Pathology

HSV-1 and HSV-2 are both transmitted through skin-to-skin contact or through contact with an infected person’s bodily fluids. HSV-1 primarily infects the areas of the mouth, throat, face, eyes, and the central nervous system, while HSV-2 primarily infects the genitals. However, both variants can infect all areas (Gupta, 2007). Transmission can also be vertical through pregnancy, and neonatal HSV infection is more severe than childhood or adult. Once infected, HSV causes blisters on the skin that are filled with active virus particles (Fig 1). Contact with these blisters carries the highest risk of transmission. However, these blisters are not permanent, lasting typically a few weeks. The infection then goes into a latent stage, where the infected person is asymptomatic. Though asymptomatic, the infected person may still transmit the virus. During the latent stage, HSV invades peripheral neurons and remains in the nuclei of these cells (Gupta, 2007). Eventually, the virus is triggered and re-infection begins, as virions move out of neuronal nuclei and out to re-infect epithelial areas. It is unknown exactly what triggers the resurgence of the virus. Interestingly, once infected other areas become resistant to infection: If an oral infection of HSV-1 occurs, then infections will not occur on the fingers (Gupta, 2007). Also, contracting one variant confers some protection against the other, as those infected with HSV-1 are somewhat resistant to HSV-2, while those infected with HSV-2 are resistant to HSV-1 (Gupta, 2007). Regardless of which strain is contracted, the immune system never eradicates the infection. Re-activations of the virus often become less severe and more infrequent over time, with some infected individuals becoming completely asymptomatic (Gupta, 2007).
Structure and Genomics

Virion Structure:
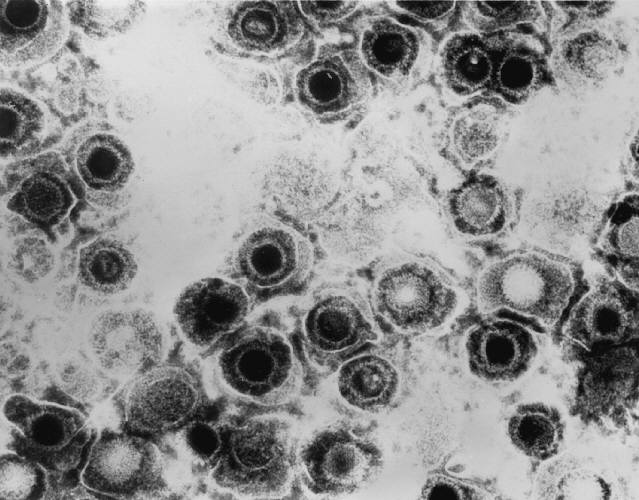
All animal herpes viruses share a similar virion structure, consisting of a protein capsid containing the viral DNA that is surrounded by a membrane envelope that is connected to the capsid by a protein tegument (Mettenleiter, 2006). HSV is a DNA based virus; it has a linear, double stranded genome. The protein capsid is in an icosahedral shape approximately 15 nm thick and 125 nm in diameter (Newcomb et al, 1996). There are five conserved proteins that comprise herpes capsids: UL19, which is the major capsid protein that is in all herpes capsids, UL18 and UL38, which are proteins that form triplexes that interact with and help to stabilize adjacent capsids, UL35, which covers all hexons, and UL6, which forms the portal through which the viral genome is injected into the nucleus (Fig 2). The capsid is connected to the membrane envelope via the viral tegument, with which these capsid proteins associate (Mettenleiter, 2006). Herpes virions have also been found to contain cellular proteins, including cytoskeletal components actin and tubulin, heat shock proteins Hsp70 and Hsp90, and annexin. However, it is unknown exactly what role these cellular proteins play and why they are recruited during replication of virions. These cellular proteins are abundant in infected cells during virion replication, and so it is uncertain if these proteins are randomly recruited as “filler material”, or whether they serve a purpose such as priming newly infected cells for synthesis of virion components (Mettenleiter, 2006).
HSV genomics:
The HSV genome is a relatively long genome, with HSV-1 and HSV-2 each encoding at least 74 genes (McGeoch et al, 2006). The herpes genome is linear, and comprised of two main regions, the unique long region (UL), and the unique short region (US). The long region contains 56 genes, while the short region contains 12 genes. Genes in both regions encode for all of the necessary structural components of the virus, including capsid, tegument, and envelope proteins, as well as genes that control viral replication processes and infective ability. Herpes utilizes the RNA Polymerase II of the infected host to transcribe its genes (McGeoch et al, 2006). The genes can be classified into immediate-early, early, and late viral genes. Immediate-early genes encode for the transcription of proteins that regulate the expression early and late genes, and these proteins are also part of the tegument that enters the cell after the capsid (McGeoch et al, 2006). Early genes are expressed after the immediate-early genes, and these genes control the biosynthesis of enzymes involved in DNA replication, as well as the production of various glycoproteins that comprise the virion envelope. The late genes primarily encode for the proteins that form the capsid and tegument and form the virion particle (McGeoch et al, 2006).
Viral Cycle
Cellular Invasion:
The virion invades the cell by fusing its envelope with the cellular envelope. This fusion of virion membrane and cellular membrane is mediated by envelope glycoproteins. Viral induced membrane fusion can be defined as three phases; Phase I has the two membranes brought into close proximity by having a viral glycoprotein bind to a cellular surface receptor, Phase II consists of the initial mixing of membrane leaflets and the formation of a hemifusion intermediate pore, Phase III has the formation and expansion of a stable fusion pore (Subramanian, 2007). HSV requires four glycoproteins to achieve fusion; glycoprotein D (gD), a complex of glycoprotein H and glycoprotein L (gHL), and glycoprotein B (gB). The presence of gD and gHL are indicated during leaflet mixing and hemifusion, while gB is present during full fusion and mixing of viral and cellular contents. A sequential model has been proposed for glycoprotein involvement in envelope fusion, with gD being necessary for Phase I as the glycoprotein that is recognized by cell surface receptors, gHL being necessary for Phase II leaflet mixing, and gB being necessary for full fusion and pore formation. All four proteins are necessary for HSV to be infectious, for without any one of the glycoproteins no cellular invasion occurs (Subramanian, 2007).
Once envelope fusion has completed, the viral tegument and capsid are transported to the nucleus of the infected cell. The tegument remains attached to the capsid, and makes its way to the nucleus by using the cytoskeleton proteins of the infected cell (Zaichick, 2013). Once the capsid reaches the nucleus, the capsid injects the linear viral DNA into the nucleus of the infected cell. This injection is done through the capsid portal, created by the formation of a twelve units of the capsid protein pUL6 (Trus et al, 2004). There is but a single portal per capsid, and it is assembled at one of the capsid’s vertices (Cardone et al, 2007).
Replication:

One of the first things that required for replication of viral DNA is the suppression of host cellular protein synthesis. This process is known as early shutoff or virion-associated host shutoff. The protein that controls this process is the UL41 gene product protein, VHS, which is a tegument protein (Matis, 2001). The activities of VHS affects multiple cellular functions, including shutoff of host protein synthesis, degradation of host mRNA. VHS remains in the cytoplasm to carry out these functions, while the viral capsid travels directly to the nucleus to inject the viral genome (Taddeo, 2006). VHS is an example of an immediate-early protein that is present in the infected cell immediately upon infection. VHS protein also interacts with cellular alpha-transinducing factor, and this interaction down-regulates VHS activity (Matis, 2001). This seems counter-intuitive, but is important because VHS does not differentiate between cellular and viral mRNA (Taddeo, 2006).
Once inside the nucleus, viral DNA reconfigures itself into a circular form, possibly involving a recombination event (Boehmer, 1997). This circularization is able to occur without protein synthesis. Evidence found in cell lines where circularization is inhibited is that circularization is required for viral DNA replication (Boehmer, 1997). This circularization allows for origin-dependent replication. There are three reported sites of origin for DNA replication within the HSV genome; oriL, which is located between the UL29 and UL30 genes, and two copies of oriS, which are located near either end of US (Boehmer, 1997). Interestingly, not all of these genes are required for replication, as deletions of either allow for viral replication. One theory is that not all that origin points are used simulatenously, that some may e active during the lytic phase while the rest are active during the reactivation from the latent infection (Boehmer, 1997). Seven viral gene products are necessary and sufficient for origin-specific viral genome replication: UL5, UL8, UL9, UL29, UL30, UL42, and UL52 (Boehmer, 1997). UL5, UL8, and UL52 encode for DNA helicase-primase, UL30 encodes for DNA polymerase with UL42 encoding for a helper protein, UL9 encodes for the origin binding protein, and UL29 encodes for the single-stranded DNA binding protein (Boehmer, 1997).
Once replicated, the DNA must cleaved and repackaged into a new capsid. As mentioned earlier, the completed capsid consists of five conserved proteins, UL6, UL18, UL19, UL35, and UL38. There are also two other proteins involved in the assembly of the capsid, two scaffolding proteins encoded by the genes UL26 and UL26.5 (Newcomb et al). UL26 is a maturational protease and UL26.5 encodes for a scaffolding protein. Capsid formation proceeds through multiple stages, first by the formation of partial capsids, then by the formation of a closed spherical capsid intermediate, and finally to the closed icosahedral capsid. The closed, spherical capsid is an unstable conformation, is a more open form than the icosahedral capsid (Newcomb et al). A conserved capsid-associated protein, UL25, is thought to play a role in envelope formation by being part of the tegument that joins the capsid to the envelope, and that it helps selectively to selectively form envelopes around capsids that contain viral DNA (as opposed to an empty capsid) (Mettenleiter, 2006).
After nucleocapsid formation, the capsid must exit the nucleus. The capsid moves towards the inner membrane of the nucleus just before the formation of the primary virion envelope. Movement within the nucleus of the capsid is facilitated by the microfilament protein actin, and contact with the inner nuclear membrane is enabled by two virally encoded proteins, UL31 and UL34 (Mettenleiter, 2006). UL31 and UL34 bind to the inner nuclear lamina and soften it by dissolving the lamin network and the chromatin layer. This allows the nucleocapsids to cross through the inner membrane layer. The movement of the capsid across the outer nuclear membrane into the cytoplasm is achieved by fusion of the primary capsid envelope with the outer nuclear membrane. The mechanism for the fusin of envelope to inner membrane is unclear, because none of the glycoproteins necessary for cellular invasion are necessary for nuclear egress (Mettenleiter, 2006).
Once the capsid has exited the nucleus, it must receive its tegument and final, secondary envelope. The tegument is actually formed at two sites, both at the capsid and at the envelope 2006). These tow subassembly sites utilize two different sets of proteins in their functions. At the capsid site, the viral rpteins encoded by UL36 and UL37, as well as proteins encoded by UL25 and US3 2006). At the envelope formation site, part of the trans-Golgi complex, glycoproteins are assembled with tegument proteins. The tegument proteins are encoded by the genes UL46, UL47, and UL49. Also key are the glycoprotein M (gM) and the protein encoded by UL11 2006). gM is an integral glycoprotein in mature virion envelopes that gathers other glycoproteins, and UL11 targets other tegument proteins to the future envelopment site. Once these subassemblies are joined, a mature virion is formed within a cellular vesicle. This vesicle then migrates to the cellular membrane and fuses with it to release the mature virion (Fig 3). An interesting facet of secondary envelopment is that these envelopes may be formed without a capsid 2006). Though these steps describe the process of HSV replication, the basic mechanisms are appear to be conserved across the Herpesviridae family 2006).
Latency and Immune Evasion:
One of the key traits of the herpes virus family is the ability to infect the host with a latent infection and remain in host cells for the life of the host. This ability is dependent on two factors; first, the virus must be able to evade the immune response of the host, and second the virus must be able to halt the lytic cycle. HSV evades immunity through interference with the major histocompatibility complex (MHC) presentation mechanism of the cell (Hill et al, 1995). HSV achieves this interference through an immediate-early protein called ICP47. ICP47 binds to a cellular protein called TAP, a transporter associated with antigen processing. ICP47 binds TAP to prevent MHC from being sent through the endoplasmic reticulum to the cell surface. Without presentation MHC, viral epitopes cannot be presented to cytotoxic T-lymphocytes, thereby allowing the virus to persist in the host by preventing lymphocyte-mediated destruction of infected cells.
Latency is achieved when the virus migrates into peripheral neurons that innervate the epithelial cells where the infection is active (Gupta, 2007). The virus invades the nucleus of the neuron, where it maintains its genome in a circular form. This circular formation is associated with nucleosomes in a chromatin configuration (Pinnoji et al, 2007). Viral genes are deactivated during latency, to prevent the lytic cycle from activating in the neuron. Neuronal factors are involved in the repression of HSV gene expression during the latent phase of the infection. One such factor is Repressor Element Silencing Transcription Factor/Neuronal Restrictive Silencer Factor (REST/NRSF) (Pinnoji et al, 2007). This factor acts on the viral gene ICP4, which is a key transactivator of HSV genes involved in the lytic cycle. By repressing the lytic cycle the viral genome simply continues to replicate within the neuronal nucleus until some trigger cause the virus to migrate back out from the neuron to the previously infected region. The mechanisms that trigger reactivation of the virus are not understood.
Capsid Transport Mechanism
The mechanism by which capsids are transported to the nucleus from the cell membrane is through the usage of the cell’s own cytoskeleton. The virus achieves this by appropriating the cellular machinery used to transport molecules along the cytoskeletal network, dyneins (Zaichick et al, 2013). Dyneins and kinesins are motor proteins that “walk” along microtubules while carrying molecules. The difference between the two is their direction of motion; kinesins move from the negative to the positive end of the microtubule, while dyneins move in the opposite direction. Current research has shown that herpesvirus tegument proteins attach to dyneins and promote retrograde motion towards the nucleus (Zaichick et al, 2013). The tegument protein that performs this action was determined to be viral protein 1/2 (VP1/2), also known as UL36, using the neuroinvasive herpesvirus, pseudorabies virus (PRV). UL36 interacts with the tegument protein UL25 for functional activation. This complex tethers the capsid to the dynein through a protein-protein interaction with a proline-rich sequence in UL36, which then carries the capsid to the nucleus for injection of the viral genome. Data from this experiment showed that mutant variants of PRV with the proline-rich sequence deletion were not as virulent as wild-type PRV, and movement of the capsid to the nucleus was impaired.
HSV as a Treatment Vector for Cancer
HSV has been an object of interest into research and development as a treatment vector for cancer. HSV has many traits that make it desirable as a treatment vector; HSV can infect a wide variety of cell types, the normal replication cycle of HSV causes cells to lyse (killing cancerous cells), The HSV genome has many genes that are non-essential to replication that can be replaced with therapeutic genes, there are already many pharmaceutical options that can be used to control against unwanted replication of the virus, and the viral genome remains as an intact plasmid within the cell nucleus which protects against unwanted insertion of viral DNA into the host genome (Varghese, 2002). HSV viruses were re-engineered to express cytotoxic genes or genes to stimulate immune response. The effectiveness of HSV as a treatment vector increased when used in conjunction with other cancer therapies such as radiation treatment and chemotherapy. Three strains of re-engineered HSV, designated G207, 1716, and NV1020, were put through Phase I clinical trials in 2002. There were no negative effects attributed to the virus. These strains were effective in the treatment of a variety of tumors in mice, including breast, prostate, colon, and pancreatic cancer, as well as rarer cancers. (Varghese, 2002).
Conclusion
Herpes viruses are incredibly common and wide infect a wide variety of organisms. Herpes simplex virus is of particular interest to humans because of its virulence and widespread infection of the human populace, its ability to leave a latent infection within the host that can evade the immune system for the life of the host and can reactivate, and its potential as a treatment vector by reengineering HSV to take advantage of the very properties that make it so virulent. Further research into HSV and other herpes family viruses will focus on indentifying more about how the virus functions and what makes it so virulent. Better understanding of this, such as Zaichick’s research on how the capsid is transported to the nucleus, will provide new avenues for targeted treatment of herpes virus infections. Simultaneously, development of HSV as a treatment vector for cancer is a promising development that could one day allow for more precise treatment of cancer than the brute force methods of chemotherapy and radiation treatment that are still used for many cancers today.
References
Edited by student of Joan Slonczewski for BIOL 238 Microbiology, 2013, Kenyon College.

{kind=link}
{kind=link}