Peroxisome Protein Import in S.cerevisiae
by Charlotte Leblang
Introduction

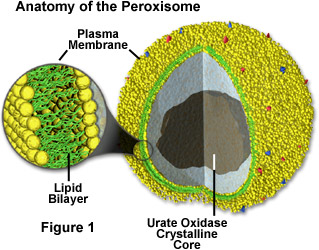
Peroxisomes (FIgure 1) are membrane-bound organelles involved in metabolic reactions and energy metabolism (1). The enzymes inside peroxisomes mostly carry out redox reactions that release hydrogen peroxide as a byproduct, which can be decomposed or converted to water by catalase (1). Catalase is also found inside the peroxisome itself. Many molecules are broken down inside the peroxisome, but fatty acid breakdown/oxidation releases the most energy to the cell. Furthermore, the peroxisome is involved in the synthesis of bile and lipids, with the help of the endoplasmic reticulum (1). Fatty acid breakdown, for example, is performed by peroxisomes and mitochondria in animal cells, but only peroxisomes in plant cells and yeast (1). Peroxisomal replication is similar to chloroplast and mitochondrial replication. They import proteins from the cytosol, and as the peroxisomes grow, the formation of new peroxisomes begins to occur. The proteins that are imported into peroxisomes are already folded; they are not imported as loose polypeptide chains like in chloroplasts and mitochondria (1).
In plants, the peroxisomes in leaves and seeds have a key role to life. In seeds, the conversion of fatty acids to carbohydrates in a cycle similar to the citric acid cycle, provides the energy necessary for plant growth (1). In the leaves of the plant, peroxisomes use a side product of the Calvin cycle (glycolate) and convert it into useable glycine and glycerate (1).
Lysosomes vs. Peroxisomes
Lysosomes mostly contain digestive enzymes to break down harmful toxins in the cell, or broken machinery such as old organelles (2). The organelle contains acid hydrolases inside, and thus needs the pH of its interior to be much lower than the cytosol. The interior pH is around 4.8 for the enzymes to function and the cytosol is about 7.2 (2). Lysosomes use both phagocytosis and endocytosis to digest debris and harmful molecules. The peroxisome also contains enzymes, but their enzymes break down more natural compounds that are not an immediate threat to the cell, such as purines, amino acids, and polyamines (3). The compounds are moved across the membrane into peroxisomes with ATP-binding cassette transporters, as opposed to phagocytosis or endocytosis (3).
S.cerevisiae as a Model

Saccharomyces cerevisiae (Figure 2) is yeast commonly used as a eukaryotic model in biology. It is commonly known as “Brewer’s” or “Baker’s” yeast because it is involved in the production of ethanol and also helps bread rise by producing gas (26). It is a good model organism because only about 34% of yeast genes that have mammal homologs have an unknown function, and only 25% of total yeast genes have unknown functions (4). Due to the vast knowledge regarding S.cerevisiae, it is the model organism for a wide majority of peroxisome and organelle research.
Ubiquination of Import Proteins

Ubiquination of peroxisomal import proteins is also important for the recycling of these proteins back into the cell cytosol. Often, when a protein becomes ubiquinated, it acts as a tag for the protein to be sent to the proteasome for degradation. This process of being sent to the proteasome also occurs with Pex18p. After aiding Pex7p with the transport of the cargo, Pex18p is ubiquinated on three residues, two lysine residues and a cysteine residue. It was found that if the cysteine is replaced or mutated in some way, then ubiquination occurs at the lysines instead. This polyubiquination tags the protein for recycling back out into the cytosol. Interestingly, however, if the residues on Pex18p are not all ubiquinated, Pex18p does not have full function or is not successfully recycled back to the cytosol. All three residues need to be ubiquinated for the recycling to occur. This is often the case for ubiquination of proteins, and this pathway is also present in other organelles in mammalian, yeast, and plant cells. Using this data, one paper proposes an “export driven import” model to cargo translocation across the peroxisomal membrane (Figure 3). It was found that Pex18p export was necessary for Pex7p import. This relates to ubiquination, because it had previously been shown for Pex5p (PTS1 signal importer) that cysteine dependent ubiquination was necessary for the cargo transport and subsequent receptor export/recycling. Thus, it is thought that perhaps the ubiquination and export of Pex18p is necessary for Pex7p to import the PTS2 tagged cargo into peroxisomes. This is a fairly new model, and perhaps it will be further studied and more widespread as more protein import systems are studied in depth.
Other Proteins in Peroxisomes

There are many groups of proteins in the peroxisomal membrane (Figure 4). Peroxisome proliferator-activated receptors are a group of proteins in peroxisomes that function as transcription factors (27). There are alpha, beta, and gamma PPARs. The alpha PPARs are expressed in liver, heart, kidney, muscle, and fat tissue. The beta PPARs are expressed in brain, skin, and fat tissues while the gamma PPARs are expressed in almost all tissue types (27). The PPARs bind directly to target DNA sequences and either upregulates or downregulates the expression of the particular DNA strand. People who carry mutations in their PPAR gamma protein suffer from PPARγ ligand resistance syndrome, which can cause insulin resistance, type 2 diabetes, hypertension, and other symptoms (7). Physically, people suffering from this mutation have low fat reserves in their limbs but a high fat content in their abdominal region (7). Gamma PPAR is also thought to have a role in cardiovascular disease, as it may control genes related to atherogenesis, the build-up of plaque in arteries (7).
Peroxisomal Biogenesis Disorders

Approximately 1 in every 50,000 people is born with a peroxisomal biogenesis disorder (Figure 5) (8). The Zellweger syndrome spectrum (ZSS) is a collection of three conditions resulting from gene mutations that cause loss of function of the peroxisomes. Infantile Refsum disease (IRD) is the least severe of the three conditions, neonatal adrenoleukodystrophy (NALD) is more severe, and Zellweger syndrome is the most devastating, often resulting in death during the infant’s first year (9). Although there are many peroxisome gene mutations involved in the cause of Zellweger syndrome spectrum, the Pex1 mutation, specifically in two alleles, is found in about 68% of people affected by ZSS (9). Pex1 is anchored to the peroxisomal membrane and helps import proteins into the cytosol. Symptoms of the Zelleweger syndrome include hypotonia (low muscle tone), mental retardation, seizures, hearing and vision loss, calcification at the end of long bones, and liver and kidney disease (10). There are also physical deformities such as a flattened face with a high forehead and broad nasal bridge (10). People with IRD and NALD experience similar symptoms to a lesser degree, and also could have a loss of learned skills and hemorrhaging in the brain (10).
The other category of peroxisome biogenesis disorders (PBDs) is the rhizomelic chondrodysplasia punctata spectrum (RCDP). RCDP differs from ZSS in the phenotypes expressed throughout infancy and childhood. Specifically, people with this disorder have a mutation in the Pex7 gene. Pex7 is involved in protein import into the peroxisome; it recognizes the PTS2 signal (11). People with RDCP also experience symptoms similar to the ZSS. They will most likely be born with cataracts in both eyes and have a small head circumference and shortened bones (12). To test if an infant or child has RCDP, the level of plasmalogens will be tested. Plasmalogens are fatty acids that are necessary for metabolic pathways in the body and their loss of function eventually leads to death (12).
Aside from the peroxisome biogenesis disorders caused by failure of peroxisomal function, there is another class of PBDs that result from single-enzyme mutations. Two of these diseases are X-linked adrenoleukodystrophy and adult Refsum disease (9). X-linked adrenoleukodystrophy affects the adrenoleukodystrophy protein (ALDP), which transports long fatty acid chains across the peroxisomal membrane (13). Adult Refsum disease is divided into two subgroups, phytanoyl-CoA hydroxylase mutations and Pex7 mutations, the former making up about 90% of cases (14). When phytanoyl-CoA hydroxylase is mutated, there is an accumulation of phytanic acid in the body. Phytanic acid is a long fatty acid chain, and the accumulation results in vision, hearing, and olfactory impairment. Individuals with peroxisomal disorders from single-enzyme mutations can live until their early twenties, unlike the early death associated with disorders caused by complete peroxisomal failure. Thus, if a small part of protein import machinery is mutated, the person survives longer than if the entirety of the machinery is mutated or missing.
Lysosomal Storage Disease
Tay-Sachs disease, a lysosome-related disease, has symptoms similar to those of peroxisomal biogenesis disorders. Symptoms of Tay-Sachs include seizures, vision and hearing loss, intellectual disability, and paralysis; and many people with this disease live only until early childhood (25). Tay-Sachs results from a mutation to the enzyme beta-hexosaminidase A, which is in lysosomes and breaks down GM2 ganglioside in the brain and spinal cord. The accumulation of GM2 ganglioside causes the degradation of neurons. This process is similar to how peroxisomal biogenesis disorders lead to the demyelination of the neurons in the brain. Martinez et.al found that stabilizing the amount of docosahexaenoic acid (DHA) in patients appeared to significantly increase myelination in the brain (15). Perhaps there is a compound that can be ingested that will aid in the breakdown of GM2 ganglioside when beta-hexosaminidase A is not present.
Peroxisome movement and division

The movement of organelles during the division of S.cerevisiae cells is mediated by class V myosins. Myo2p is the motor for the membrane bound organelles in S.cerevisiae, including the golgi apparatus, the peroxisome, vacuoles, and mitochondria (16). Each organelle was proposed to have its own binding site for Myo2p, and on peroxisomes it is Inp2p. Since Myo2p interacts with different organelles at different times during the cell cycle, the levels of Inp2p in peroxisomes fluctuate (16). They are at their highest level while being transported into the daughter cell, and once half have been moved, the levels begin to decrease before the final step in cell division, cytokinesis (16). There have not been many studies regarding the Inp2p-Myo2p interaction, but it has been found that mutations in the Myo2p tail lead to incomplete peroxisome inheritance (16). If this causes the amount of peroxisomes in the cell to be low, there is another set of proteins that can assist in forming new peroxisomes.
A set of peroxisomal membrane proteins (PMPs) has been found to be an integral part of creating new peroxisomes in a peroxisome-deficient cell (17). They insert into the endoplasmic reticulum membrane with other import complexes, Sec61p and Get3p, and then organize themselves into a useful form and are transferred to peroxisomes (17). Each protein was found to have different exit points from the endoplasmic reticulum, and then move to the peroxisomes to import PTS1 and PTS2 proteins to form the new and functioning peroxisome (17). This pathway, moving from the endoplasmic reticulum to the new peroxisome, makes sense because the peroxisomes cannot divide indefinitely, and this pathway sustains the continuous supply of peroxisomes to the cell. Other than large sets of proteins working together, there are some proteins that work alone for very specific functions.
Rho1p is involved in the reorganization of actin into peroxisomes (18). Rho1p mutants have been shown to produce fewer and smaller peroxisomes than the wild-type, thus Rho1p has been hypothesized to be involved in the fusion of peroxisome precursor proteins (18). Pex25p has been shown to also induce growth disorders in peroxisomes: the deletion of Pex25p or a protein in the same family (Pex11p or Pex27p) caused peroxisomes to become dense and enlarged: deletion of Pex31 and Pex32 also resulted in enlarged peroxisomes (18). Deletion of Pex28p or Pex29p, however, caused a large number of small peroxisomes that clumped together, so perhaps these proteins are involved in peroxisome separation (18). Deletion of Pex23p resulted in only immature peroxisomes, and deletion of Pex30p caused an overall increase in the number of peroxisomes (18). Interestingly, the deletion of Pex30p, Pex31p, and Pex32p caused a five-fold increase in normal peroxisomes (18). The interaction of Pex16p with acyl-CoA oxidase (Aox) is important in the replication of peroxisomes; the interaction causes a curve in the peroxisomal membrane to being proliferation (18). It is also thought, however, that peroxisome proliferation could be related to the overexpression of some transcription factors, instead of depending on metabolism rates. Another known protein involved in peroxisome proliferation, however, is a dynamin-related protein (DRP), and these proteins are also involved in chloroplast and mitochondrial division (18).

UPS in Chloroplasts and Mitochondria
The ubiquitin-proteasome system is similar in chloroplasts. As in peroxisomes, the chloroplast protein import machinery also uses ubiquination to recycle and degrade proteins (Figure 6). Ling et al. used SP1, a ubiquitin ligase phenotypically similar to E3 enzymes, to discover the relationship between ubiquitin and the chloroplast TOC (23). E3 enzymes help attach ubiquitin to proteins, and are represented by the RING finger complex in both chloroplasts and peroxisomes. The SP1 protein was found to associate with TOC complexes and mediate their ubiquination and thus degradation (23). The authors found that when SP1 was overexpressed, TOC protein levels decreased, indicating SP1 ubiquination controlled the TOC levels in chloroplasts. This indicates that SP1 is a mechanism for the regulation of biosynthesis of the chloroplast, because it can control the levels of all TOC machinery. Similar in peroxisomes, the level of Pex18p depends on how often it is ubiquinated, and Pex18p is an integral part for importing proteins across the peroxisomal membrane.
The ubiquitin-proteasome system (UPS) of the mitochondrial intermembrane space (IMS) also shares similarity to peroxisomes (Figure 7). In a study by Bragoszewski et al., it was found that the UPS was not just involved in removing failed proteins, but also involved in the biogenesis of IMS proteins (24). IMS proteins are involved in many metabolic functions, including mitochondrial biogenesis (24). The MIA pathway folds the proteins imported into the IMS by adding disulfide bonds and trapping them in the space. The authors first found that when the MIA pathway was knocked out, there was no accumulation of proteins in the IMS so they must be being degraded by the proteasome. It was then found that when proteasome activity was inhibited, the IMS proteins accumulated. Conclusively, it was shown that the proteasome was inhibited before the proteins imported into the IMS, thus allowing MIA substrates to accumulate. The function of the MIA substrates is to either degrade or import proteins, therefore they regulate the IMS proteins (24). Since the UPS controls the level of MIA substrates and subsequently IMS proteins, the UPS is part of the regulation of mitochondrial biogenesis at the post-translational level. This is similar to peroxisomes because the UPS controls the levels of protein import machinery, thus it regulates all proteins being transported into peroxisomes, including biogenesis proteins.
Relevance to More Common Diseases
X-linked adrenoleukodystrophy, a peroxisomal biogenesis disorder mentioned above, hinders an ATP-binding transporter in the membrane of peroxisomes. The transporter is responsible for moving very long chain fatty acids into the peroxisome so they can be processed. The impairment of this transporter thus causes a large build up of very long chain fatty acids in the brain. This results in inflammation in the brain and therefore demyelination of neurons. Myelin is made up of docosahexaenoic acid (DHA), an omega-3 fatty acid. A study recently found that the insertion of DHA into patients who have X-linked adrenoleukodystrophy and demyelination, increased myelination in the brain (15). Multiple sclerosis affects millions of people in the world, and a symptom is also demyelination. The treatments for MS are uncomfortable and costly, thus it would be beneficial if this DHA treatment worked for MS as well. Since it is also a demyelination disease as a result of inflammation in the brain, ideas for treatments may come from other diseases. Conclusively, it is important to research less common diseases, because sometimes the symptoms and causes of diseases can be similar (as shown about with Tay-Sachs disease).
Conclusions
Peroxisomes are membrane-bound organelles found in plants and animals that are involved in energy metabolism and oxidative reactions. Although some aspects of peroxisomes are similar to lysosomes, it is apparent that these organelles have distinct functions. Much of the research being done on peroxisomes uses S.cerevisiae as a model organism, because a lot is known regarding its genes and mammalian homologs. Ubiquination seems to be an integral part to the import of proteins into peroxisomes, and then recycling the receptors back into the cytosol. If the mechanistic proteins are impaired, they can cause peroxisomal biogenesis disorders, which cause mental disabilities and early death in most people. There are differing degrees of peroxisomal biogenesis disorders, with Zellweger being the most fatal. The many other proteins embedded in the peroxisomal membrane also have unique functions, and impairment can lead to diseases such as type-2 diabetes. Other protein in the peroxisome have been characterized to aid in peroxisomal movement and division, and some have also been found in chloroplasts and mitochondria. Protein import into chloroplasts and mitochondria also has similarities to protein import into peroxisomes. Knowledge regarding protein import, peroxisome division and movement, biogenesis disorders, and signaling, will be useful to eventually find cures for life-threatening diseases, even if they're not peroxisome related.
References
(1)Cooper GM. The Cell: A Molecular Approach. 2nd edition. Sunderland (MA): Sinauer Associates; 2000. Peroxisomes. Available from: http://www.ncbi.nlm.nih.gov/books/NBK9930/
(2)"Difference and Similarity between Lysosomes and Peroxisomes | Biology."Biology Discussion. N.p., 04 Mar. 2014. Web. 17 Nov. 2015.
(3)Visser, W. F., van Roermund, C. W. T., Ijlst, L., Waterham, H. R., & Wanders, R. J. A. (2007). Metabolite transport across the peroxisomal membrane.Biochemical Journal, 401(Pt 2), 365–375.
(4)Botstein, D., Chervitz, S. A., & Cherry, J. M. (1997). Yeast as a Model Organism. Science (New York, N.Y.), 277(5330), 1259–1260.
(5)Otera, H., Setoguchi, K., Hamasaki, M., Kumashiro, T., Shimizu, N., & Fujiki, Y. (2002). Peroxisomal Targeting Signal Receptor Pex5p Interacts with Cargoes and Import Machinery Components in a Spatiotemporally Differentiated Manner: Conserved Pex5p WXXXF/Y Motifs Are Critical for Matrix Protein Import. Molecular and Cellular Biology, 22(6), 1639–1655.
(6)Andrzej Jankowski, Jae Hong Kim, Richard F. Collins, Richard Daneman, Paul Walton, and Sergio Grinstein. MEMBRANE TRANSPORT STRUCTURE FUNCTION AND BIOGENESIS:In Situ Measurements of the pH of Mammalian Peroxisomes Using the Fluorescent Protein pHluorin (2001). J. Biol. Chem. 2001 276: 48748-48753.
(7)Meirhaeghe, A., & Amouyel, P. (2004). Impact of genetic variation of PPARγ in humans. Molecular Genetics and Metabolism, 83(1–2), 93-102.
(8)Zellweger spectrum disorder. (2015). Retrieved 9/15, 2015, from http://ghr.nlm.nih.gov/condition/zellweger-spectrum-disorder
(9)Steinberg, S., Raymond, G., Braverman, N. & Moser, A. (2012). Peroxisome biogenesis disorders, zellweger syndrome spectrum. Retrieved 9/15, 2015, from http://www.ncbi.nlm.nih.gov/books/NBK1448/
(10)"Diseases." Zellweger Syndrome Spectrum. N.p., n.d. Web. 17 Nov. 2015.
(11)Braverman, N., Moser, A. & Steinberg, S. (2012). Rhizomelic chondrodysplasia punctata type 1. Retrieved 9/15, 2015, from http://www.ncbi.nlm.nih.gov/books/NBK1270/
(12)"Rhizomelic Chondrodysplasia Punctata (RCDP)." Genetic Disorders UK -. N.p., n.d. Web. 17 Nov. 2015.
(13)Steinberg, S., Moser, A. & Raymond, G. (2015). X-linked adrenoleukodystrophy. Retrieved 9/15, 2015, from http://www.ncbi.nlm.nih.gov/books/NBK1315/
(14)Wanders, R., Waterham, H. & Leroy, B. (2015). Refsum disease. Retrieved 9/15, 2015, from http://www.ncbi.nlm.nih.gov/books/NBK1353/
(15)Martínez, M., Vázquez, E., García-Silva, M. T., Manzanares, J., Bertran, J. M., Castelló, F., et al. (2000). Therapeutic effects of docosahexaenoic acid ethyl ester in patients with generalized peroxisomal disorders.The American Journal of Clinical Nutrition, 71(1), 376s-385s.
(16)Fagarasanu, A., Mast, F., Knoblach, B., Jin, Y., Brunner, M., Logan, M., Glover, M., Eitzen, G., Aitchison, J., Weisman, L., Rachubinski, R. Myosin-driven peroxisome partitioning in S. cerevisiae (2009). The Journal of Cell Biology, 186(4), 541-544.
(17)Van der Zand, A., Braakman, I., & Tabak, H. F. (2010). Peroxisomal Membrane Proteins Insert into the Endoplasmic Reticulum. Molecular Biology of the Cell,21(12), 2057–2065.
(18)Yan, M., Rayapuram, N., & Subramani, S. (2005). The control of peroxisome number and size during division and proliferation. Current Opinion in Cell Biology, 17(4), 376-383.
(19)Soll, J., & Schleiff, E. (2004). Protein import into chloroplasts. Nature Reviews; Molecular Cell Biology, 5, 198-208.
(20)Platta, H. W., & Erdmann, R. (2007). The peroxisomal protein import machinery. FEBS Letters, 581(15), 2811-2819.
(21)Wiedemann, N., Frazier, A. E., & Pfanner, N. (2004). The protein import machinery of mitochondria. Journal of Biological Chemistry, 279(15), 14473-14476.
(22)Fewell, S., & Brodsky, J. (2000). Entry into the endoplasmic reticulum: Protein translocation, folding and quality control. Retrieved 9/15, 2015, from http://www.ncbi.nlm.nih.gov/books/NBK6210/
(23)Ling, Q., Huang, W., Baldwin, A., Jarvis, P. (2012). Chloroplast Biogenesis Is Regulated by Direct Action of the Ubiquitin-Proteasome System. Science, 338, 655-659.
(24)Bragoszewski, P., Gornicka, A., Sztolsztener, M. E., & Chacinska, A. (2013). The Ubiquitin-Proteasome System Regulates Mitochondrial Intermembrane Space Proteins. Molecular and Cellular Biology, 33(11), 2136–2148.
(25)"Tay-Sachs Disease." Genetics Home Reference. N.p., n.d. Web. 17 Nov. 2015.
(26) "Saccharomyces Cerevisiae." Saccharomyces Cerevisiae. N.p., n.d. Web. 18 Nov. 2015.
(27)Wikipedia. Peroxisome proliferator-activated receptor. Wikimedia Foundation, n.d. Web. 18 Nov. 2015.

{kind=link}
{kind=link}
{kind=link}
{kind=link}